![バイオインフォ 道場 [bioinfo-Dojo]](https://bioinfo-dojo.net/wp-content/uploads/2016/03/some_object_luca-bravo-alS7ewQ41M8-unsplash.jpg)

samtoolsを使ったVariant callingについてまとめます。

解析作業の流れ
- マッピング
- Calling SNPs
- フィルタリング
- アノテーション付与
主なツール
Calling SNPsには、samtoolsを使いました。samtools
$ samtools --version samtools 1.3(Using htslib 1.3) $ bcftools --version bcftools 1.3(Using htslib 1.3)
フィルタリングやSNPdbの情報付与には、vcftoolsのvcf-annotateを使いました。vcftools
$ vcftools -h VCFtools (0.1.15)
Calling SNPsについて
マッピング
マッピングは、bowtie2やbwaを使って行います。今回は、マッピングが完了したbamファイルから使います。ユニークにマッピングされたリードの抽出・重複リードの除去は、必要に応じて行います。
ユニークリードの抽出・重複リードの除去
$ samtools view -h myfile.bam | grep -v XS:i: | samtools view -bS - > myfile.sorted_uniq.bam $ samtools rmdup myfile.sorted.bam myfile.sorted.nodups.bam
Calling SNPs
samtools mpileupコマンドの結果をbcftoolsのコマンドにパイプ連結してSNPsをコールします。特に一連の作業で、bcftoolsで「view」コマンドを使っていましたが、最新版(1.3.1)では「call」を使います。bcftoolsのサイトでは、「call…SNP/indel calling(former “view”)」と説明されています。
$ samtools mpileup -uf ref.fa myfile.sorted_unique.bam | bcftools call -O b -v -c ->| myfile.sorted_unique.raw.bcf
主なオプション
- -u generate uncompressed VCF/BCF output
- -f faidx indexed reference sequence file
- -B disable BAQ (per-Base Alignment Quality) <必要に応じて利用>
フィルタリング
Calling SNPsの結果に対してフィルタリングを行います。結果として得られたVCFファイルに対して、VCFtoolsの1つvcf-annotateでフィルタリング情報を付与します。「Strand Bias」や「Base Quality Bias」、「read depth」などの情報を付与・チェックを行い、フィルタリング情報として活用することができます。尚、全てクリアすると「PASS」になるようです。
$ bcftools view myfile.sorted_unique.raw.bcf | vcf-annotate -f + > myfile.sorted_unique.Tag.vcf
主なオプション
- + Apply all filters with default values (can be overriden, see the example below).
Filters : StrandBias, BaseQualBias, MapQualBias, EndDistBias, MinAB …
- –fill-type
Annotate INFO/TYPE with snp,del,ins,mnp,complex
- -n, –normalize-alleles
Make REF and ALT alleles more compact if possible<結果をコンパクト表示>
アノテーション
アノテーション付与1
Calling SNPsの結果として得られたvcfファイルに対して、分析やアノテーション情報を付けることのできる便利なwebツールがあります。
使い方は、とても簡単です。生物種を選んで、vcfファイルをupload、付与したい情報をチェックして「Run」。結果も、テキスト形式やVCF形式でダウンロードすることができます。今回、1,000行程度のvcfファイルをアップロードしましたが、数分程度で結果が得られました(環境によります)。尚、アップロードの上限は50Mのようです。ヘルプ&ドキュメント・FAQもあるので参考にしてみて下さい。
Variant Effect Predictor
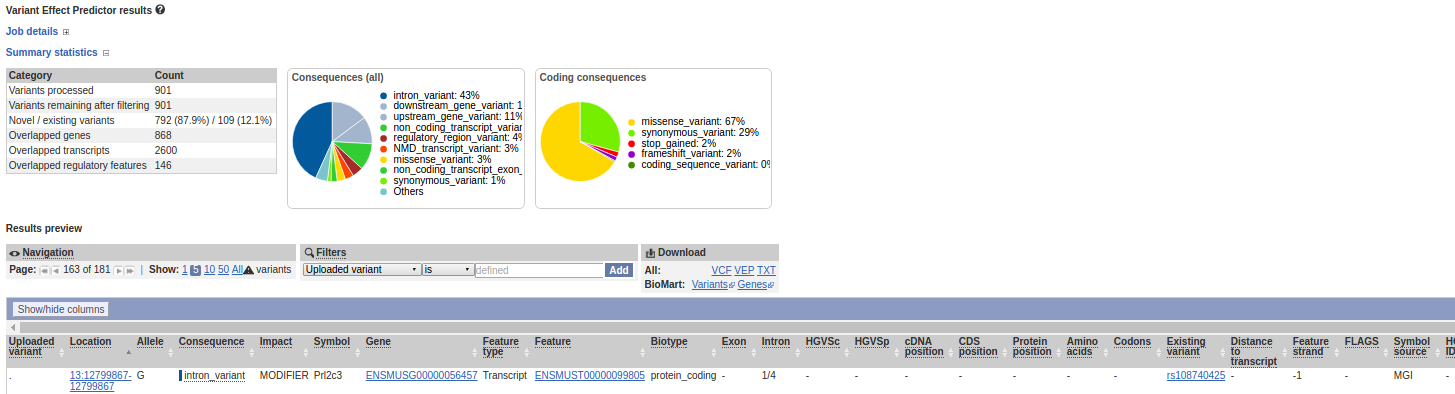
アノテーション付与2(SNPdb情報)
SNPdb情報を付与します。ツールは、「フィルタリング」でも使ったvcf-annotateを使います。先の「アノテーション付与1」で「existing variant」として情報付与されるので、あえて個別に付けたいときだけ参考にしていただければ良いと思います。この場合、準備も少し必要です。
SNPdbファイルの準備
SNPdbの情報が定義されたファイルを準備します。vcfやbed形式のファイルが便利です。今回はマウスのSNPdb情報として、Mouse Genomes Project | Sanger Instituteを参考にしました(ftp)。SNPs_Indelsに関するファイルがvcfフォーマットで提供されています。
SNPdbファイルの整形
vcf-annotateを使ってknown SNPsの情報を付与するために、ファイルを整形します。タブ区切りで、chr, start, end, idという形式にします。
chr1 1360 1361 rs000000001
vcfファイルは、下記のように少し複雑な形式をしています。
#ヘッダー情報 #.... #CHROM POS ID REF ALT QUAL FILTER INFO FORMAT ... 1 3001490 rs31521921 C A 243.50 PASS AC1=1;AC=22; ... ...
今回使うのは、先頭の数カラムなのでawkなどの簡単なスクリプトで処理して整形します。また、続けてtabixでインデックス処理を行います。
$ zcat origin.vcf.gz | awk '{OFS="\t"; if (!/^#/){print "chr"$1,$2-1,$2,$3}}' > SNP.bed
$ bgzip SNP.bed
$ tabix -p bed SNP.bed.gz
SNPdb情報付与
vcf-annotateの「-a –annotations」オプションを使って、準備したbed形式のファイルからSNPsの情報を付与します。
bcftools view myfile.sorted_unique.raw.bcf | vcf-annotate -a SNP.bed.gz -c CHROM,FROM,TO,INFO/SNP_ID -d key=INFO,ID=SNP_ID,Number1,Type=Integer,Description='SNP site' > myfile.sorted_unique.snp.vcf
出力として得られたvcfファイルのINFOにSNP_IDとしてSNPsの情報が付与されます。
論文・データ解析の支援
個別に直接相談できるのが良いと、たくさんの方にご利用頂いています。

関連記事1

関連記事2








